Up to MicrobesOnline
FastHMM and FastBLAST are fast heuristics to replace HMM search, InterProScan, and all-versus-all BLAST. FastHMM uses PSI-BLAST to quickly select likely members of the family and then uses HMMer to confirm those hits. FastBLAST relies on alignments of proteins to known families from FastHMM and from rpsblast against COG. FastBLAST uses these alignments to avoid most of the work of all-versus-all BLAST. FastBLAST further reduces the work by clustering similar sequences. FastBLAST runs in two stages: the first stage identifies additional families and aligns them, and the second stage quickly identifies the homologs of a query sequence, based on the alignments of the families. FastHMM and the first stage of FastBLAST analyzed 6.5 million protein sequences from Genbank NR in about a week on a cluster of 200 CPUs.
More information:Downloads and instructions are available from:
FastHMM and FastBLAST have been tested on 64-bit x86 Linux machines with Perl 5.8.5, HMMer 2.3.2, BLAST 2.2.14 or 2.2.17, MUSCLE 3.52, and CD-HIT 2006 or 2007. We do not know of any reason why they would not work on other platforms or with other versions of these packages, but we have not tested that. If you run them on a non-Unix machine, you will also need to install Unix-like tools (e.g. cygwin).
FastHMM requires at least 15 gigabytes of free disk space if you download and install all seven family databases. You'll need even more disk space for the output. For example, if you want to run both FastHMM and FastBLAST on NR (currently 2.2 GB), we recommend that you have 250 GB of free disk space.
Before you install FastHMM/FastBLAST, please make sure you have the following software (you'll probably already have most of it):
FastHMM uses profile Hidden Markov Models (profile HMMs) to find instances of known families and domains within a protein sequence database. HMMs are very sensitive but also rather slow, so to speed up the identification, FastHMM uses BLAST with gaps, position-specific weights, and sensitive settings to quickly find candidate members of each family. Given these candidates, FastHMM uses HMMer to select the true family members.
FastHMM works with all seven databases of HMMs that are part of InterPro. FastHMM runs up to 60-times faster than InterProScan on genome-scale inputs and gives very similar results.
The databases of HMMs are:
For FastHMM's cutoffs and rules for assigning domains, see here. You can modify these to some extent by modifying the $FASTHMM_DIR/conf/fastHmm.conf file.
Because FastHMM uses the hmmsearch program of HMMer to validate the results from BLAST's blastpgp program, there are no false positives. To increase sensitivity on the non-redundant databases (Pfam, Pirsf, SMART, and TIGRFAM), FastHMM uses hmmsearch on weak or "hard" models. For these weak families, sequences in the seed alignment are often missed by blastpgp (or even by hmmsearch).
With default settings, the sensitivity is 94-99%. The lowest sensitivity is for Superfam (94%), but Superfam includes redundant models, which reduces the importance of the misses for annotation. Most of these missed hits are very weak: for Superfam, their median score is 12 bits, and their median e-value is 0.002.
The speedup of FastHMM increases with the size of the input -- FastHMM issues several commands for each of the tens of thousands of HMMs in the databases, so it has considerable "fixed costs." Thus, we do not recommend the use of FastHMM on small numbers of sequences. For a subset of 3% of NR, FastHMM is about 33 times faster than hmmsearch 2.3, and hmmsearch is itself about twice as fast as hmmpfam (which is the basis for InterPro). FastHMM is designed to use minimal memory even on large datasets, so you need not chop the input database into pieces. Indeed, doing so will degrade performance.
Once FastHMM is installed, you can run it as follows:
export FASTHMM_DIR=~/fasthmm $FASTHMM_DIR/bin/fastHmm.pl -i input_database -t all -j nCPUs -o output_directory
The input database must be both a fasta-format file and a valid BLASTp database. You can use the -f argument if you want fastHmm.pl to make the BLASTp database for you. If you're planning to run FastBLAST after running FastHMM, you should run cleanFasta.pl first (see below).
The -j and -o arguments are optional: -j defaults to 2, and -o defaults to the current directory. For each of the databases of HMMs, produces two files: raw output in output_directory/results.input.hmmdb.hmmhits and processed output output_directory/results.input.hmmdb.domains. The hmmhits files contain the raw hits to the models that met the thresholds, and the domains files have a non-redundant subset of those results, which corresponds roughly to predicted domains. These files are described in more detail in the output section.
fastHmm.pl writes many temporary files to /tmp and also to the output directory (-o, which defaults to the current working directory). Do not run multiple instances of fastHmm.pl into the same output directory at the same time.
To run fastHmm.pl on a single database of HMMs, specify gene3d, panther, pfam, pirsf, smart, superfam, or tigrfam with the -t argument. By default, fastHmm.pl looks for these databases of HMMs in $FASTHMM_DIR/db. If want it to look somewhere else, use the -d option. If you want to use FastHMM with your own collection of models, see the documentation in $FASTHMM_DIR/db/README.
To run hmmsearch exhaustively for every model in the database, use the -ha option. This is useful for testing, but is very slow.
fastHmm.pl has many more options -- for information about them, run fastHmm.pl without any arguments.
To run FastHMM on a cluster, use the -b option to prepare sets of commands. You must use fully specified paths and you must use the -o option. The sets of commands that it creates can run independently of each other, so you can submit them to a cluster, using whatever scheduler you choose. Then you run fastHmm.pl again with -m to "merge" the results. For example, to issue jobs as batches of 100 models, and to use 2 CPUs in each issued job, and to send the results to the fasthmm subdirectory:
export FASTHMM_DIR=~/fasthmm mkdir fasthmm $FASTHMM_DIR/bin/fastHmm.pl -t all -j 2 -i `pwd`/input.faa -b 100 -f -o `pwd`/fasthmm > fasthmm.cmds # submit each line in fasthmm.cmds as a cluster job, and wait for them to finish $FASTHMM_DIR/bin/fastHmm.pl -t all -j 2 -i `pwd`/Aful.faa -b 100 -f -o `pwd`/fasthmm -m # Post-processing to get InterProScan-like domain assignments $FASTHMM_DIR/fastHmm.pl -t all -j 2 -i `pwd`/Aful.faa -b 100 -f -o/fasthmm `pwd` -p # submit each line in result.Aful.panther.hmmhits.cmds.1 as a cluster job, and wait for them to finish # run the commands in result.Aful.panther.hmmhits.cmds.2 (will not take long)
The second set of cluster jobs, beginning with fastHmm.pl -p, is only required for making the domains calls for PANTHER, and can be omitted if you're running another database or do not need the domains calls for PANTHER. To check if any jobs failed, check the log files for "FATAL". (FastHMM sends progress information to standard out and any errors to standard error.)
For each HMM database, FastHMM produces a results.input.hmmdb.hmmhits file and a results.input.hmmdb.domains file. The hmmhits files include all hits to the HMM that meet the cutoffs recommended by the curators of the HMM (e.g., for Pfam, the gathering cutoff). The domains files include a non-redundant subset of those hits, which corresponds roughly to the output of InterProScan. Both files are tab-delimited with the following fields:
FastBLAST is a replacement for all-versus-all BLAST. As its input, it takes a BLAST protein sequence database, alignments of those genes to known families (e.g. from FastHMM or from reverse PSI-BLAST). FastBLAST runs in two stages: the first stage is a large parallel computation that identifies additional families and aligns them. On NR, the first stage is about 25 times faster than all-versus-all BLAST. The second stage uses the alignments of the families to quickly identify homologs for a gene from the database. By default, the second stage tries to identify the top N/2,000 hits, where N is the number of sequences in the database. For the average protein in NR, the second stage is 8 times faster than BLAST and identifies virtually all of the top hits (98% of the top 3,250 hits above 70 bits).
An overview of how FastBLAST works:
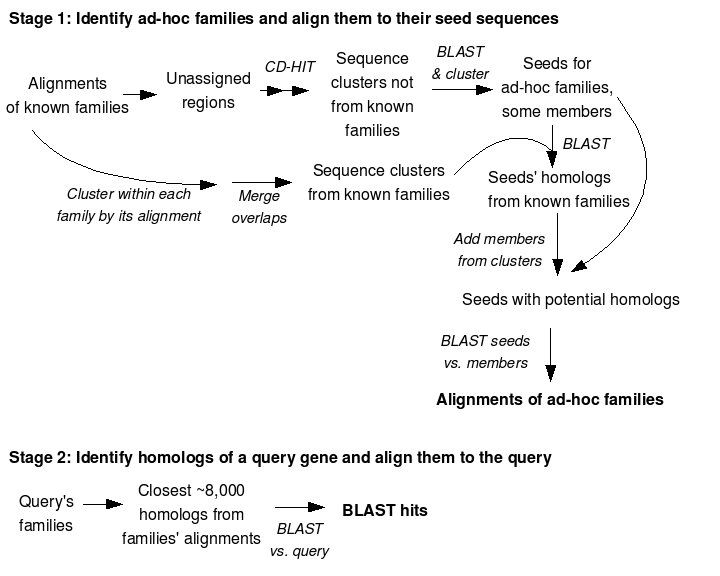
FastBLAST requires a protein sequence database in fasta format. FastBLAST uses several other tools with odd constraints on the names of the genes in the fasta file, so we recommend using cleanFasta.pl to ensure that they are compatible with FastBLAST, and making a new BLAST database, e.g.
export FASTHMM_DIR=~/fasthmm $FASTHMM_DIR/bin/cleanFasta.pl -faa mydb.faa -out clean.faa $FASTHMM_DIR/bin/formatdb -i clean.faa -p T -o T
cleanFasta.pl will create a tab-delimited file, clean.map, that describes the mapping between the cleaned identifiers and the original identifiers.
The other inputs to FastBLAST are the alignments of your protein sequences to known families. We recommend using the FastHMM and reverse PSI-BLAST against COG to generate these alignments. Running FastHMM is described above. FastBLAST only needs the raw hmmhits (the *.hmmhits files), not the domain assignments. To run reverse PSI-BLAST in parallel and get output in FastBLAST format, use the COGHits.pl tool, e.g.
export FASTHMM_DIR=~/fasthmm $FASTHMM_DIR/fasthmm/bin/COGHits.pl -faa nr.faa -pieces 500 # issue the commands in cogrps.cmds to your cluster, or just run them # then run cogrps.finish.cmd, e.g. source cogrps.finish.cmd
will create the result.nr.cogrps file.
If you want to use exhaustive HMM search instead of FastHMM, you can still use FastHMM, but run it with the -ha option. If you already have hmmsearch output files, you can use the parseHmmAlignments.pl script included with FastHMM/FastBLAST to convert the output. You can't easily use InterProScan output as the starting point for FastBLAST because it's not straightforward to get from there to alignments to models: information about both the alignment and the model is removed during InterProScan's processing. If you create the input files yourself, you need to make sure that each input file is sorted by domainId and that no domainId appears in more than one of the input files.
Once you have cleaned your protein database and created alignments, use fastBlastPrepare.pl to verify that the input files are OK and to create a makefile, and then run GNU make or qmake to run those jobs, e.g.
export FASTHMM_DIR=~/fasthmm $FASTHMM_DIR/bin/fastBlastPrepare.pl -pieces 1000 -faa clean.faa fasthmm/result.clean.*.hmmhits result.clean.cogrps # on a multi-processor machine with 4 CPUs make -j 4 -f Makefile.fastblast >& fastblastlog & # or, on a SunGridEngine cluster with 200 CPUs qmake -cwd -v PATH,FASTHMM_DIR -- -j 200 -f Makefile.fastblast < /dev/null >& fastblastlog &
fastBlastPrepare.pl creates a sub-directory "fb" to store the intermediate results. Use -pieces 1000 if you have a large cluster. On a small cluster or on a machine with a few CPUs, set -pieces to a few times the number of CPUs you have.
By default, FastBLAST stores temporary files in /tmp. Some of the temporary files are quite large: for a database of 2.2 GB of amino acids, you need about 60 GB of space in /tmp. If your /tmp filesystem does not have enough free space, use the -T option to fastBlastPrepare.pl to specify a different temporary directory (ideally one on a local disk), or edit the TMPDIR line in Makefile.fastblast.
qmake will generate a large number of jobs, most of them named "sh". qmake will report something like
qmake: *** cannot determine architecture from environment variable SGE_ARCHwhich can safely be ignored. Any other *** statements from qmake errors.
The -v argument to qmake sets environment variables that should be passed to the jobs that it issues. If you are not using the default perl installation on your cluster, you may need to pass the perl library directory as well, e.g.
-v PATH,FASTHMM_DIR,PERL5LIB
If you have a cluster, but it does not support qmake or other forms of cluster-based make, then you can still use make to coordinate your jobs. You should run make on intermediate targets or run make -n to generate lists of jobs that you submit to the cluster, like this:
make -f Makefile.fastblast -j 4 reduce1 make -f Makefile.fastblast -n blast1 > cmdlist1 # run the jobs in cmdlist1 to your cluster make -f Makefile.fastblast -j 4 maskother make -f Makefile.fastblast -n blast2 > cmdlist2 # run the jobs in cmdlist2 to your cluster make -f Makefile.fastblast -j 4 expand make -f Makefile.fastblast -n blast3 > cmdlist3 # run the jobs in cmdlist3 to your cluster make -f Makefile.fastblast all
Most of the computational effort in FastBLAST is in first two rounds of blast jobs. ("blast1" actually includes the jobs to reduce the known families, which is also a significant amount of work.) So running the other steps on a machine with a few CPUs won't actually slow things down that much.
If any of the FastBLAST jobs crash, or if your cluster crashes, you can just re-run make and it will pick up where it left off. Incomplete output files shouldn't be a problem, because most of FastBLAST's jobs don't create the output file until they are finished (they write to a temporary file first). The only exception to this rule are the BLAST formatdb jobs. If you would prefer to start over, you can use
make -f Makefile.fastblast clean
to delete all of the output files and intermediate files. (It removes all files named fb.* and the contents of the fb/ subdirectory.)
The output from the first stage of FastBLAST is the families for each gene and the alignments for each family, for both the families given to FastBLAST and the additional families identified by FastBLAST. These files are in fb.all.align and fb.all.domains.bygene. FastBLAST also creates auxilliary files (fb.all.nseq and fb.all.*.seek.db) that are required by the second stage of FastBLAST. Finally, the first stage produces auxilliary files (fb.other.masked.faa, fb.domains.merged.faa, fb.other.reduced.comb.clstr.byexemplar, fb.domain.clstr.byexemplar) that are not needed for the second stage of FastBLAST, but that you will need if you wish to add new sequences to your database with fbmerge.make. To save space, you can delete the files in the fb/ subdirectory.
The settings for the first stage of FastBLAST are in the $FASTHMM_DIR/conf/fastBlastMake.conf file, and are copied from there into Makefile.fastblast by fastBlastPrepare.pl. Modify the makefile to tweak a particular run of FastBLAST, or modify the conf file to modify future makefiles created by fastBlastPrepare.pl. See $FASTHMM_DIR/lib/fastblast.make, which is included by the Makefile.fastblast files, to see how these settings are used.
Once the first stage of FastBLAST is complete and new families have been identified and aligned, the topHomologs.pl script can quickly identify homologs for a query gene, e.g.
export FASTHMM_DIR=~/fasthmm $FASTHMM_DIR/bin/topHomologs.pl -i . -f clean.faa -l queryAccession > hits
The output of topHomologs.pl is the same format as that of BLAST and is controlled by the -m option. BLAST will probably put "lcl|" on the front of every accession in the output.
The fb.all.domains.bygene file from the first stage is in the same format produced by FastHMM, but is sorted by gene id instead of by domain id. The ad-hoc domains are named fb.SeedAccession.begin.end; the other domain ids are identical to the input.
The fb.all.align file is tab-delimited with the fields domainId, geneId, alignedSequence, geneBegin, geneEnd, score, evalue. For ad-hoc domains, the score and evalue are from BLAST. The alignedSequence does not contain all positions from the gene, only those that align to the family's model or (for ad-hoc families) to the seed sequence.
If you want programmatic access to this data, use the FastBLAST.pm module in $FASTHMM_DIR/lib.
FastBLAST includes a "merge" tool that you can use to add new sequences to your FastBLAST database. To do so, you first need to run FastHMM and FastBLAST on your new sequences. For both sets of sequences, you must use the same versions of the databases of known families. Then, you can use fbmerge.make to construct a new FastBLAST database, in the current directory, that contains sequences from both databases:
export FASTHMM_DIR=~/fasthmm mkdir mergedirectory cd mergedirectory
and then run either make (if you have a single parallel computer)
make -j 4 NPIECES=100 TMPDIR=/tmp DIR1=../fb.original FAA1=../fb.original/original.faa FAA2=../fb.extra/extra.faa DIR2=../fb.extra/extra.faa -f $FASTHMM_DIR/lib/fbmerge.make >& fbmerge.log &
or qmake (if you have SunGridEngine cluster):
qmake -cwd -v PATH,FASTHMM_DIR -- -j 100 NPIECES=100 TMPDIR=/tmp DIR1=../fb.original FAA1=../fb.original/original.faa FAA2=../fb.extra/extra.faa DIR2=../fb.extra/extra.faa -f $FASTHMM_DIR/lib/fbmerge.make < /dev/null >& fbmerge.log &
The -j option sets the number of threads or jobs to run, and the = arguments are the settings for fbmerge.make:
fbmerge.make produces a combined blast database in combined.faa (in the current directory). You can use that file, and the fb.* files in the current directory (i.e., in mergedirectory), to run topHomologs.pl. Once you have a merged FastBLAST database and have verified that it is correct (e.g., by running some test topHomologs.pl jobs), you can delete the old FastBLAST databases.
Make sure that you use the same version of FastHMM and the same databases of known families for both the original data-set and the new data-set. Also, since fbmerge.make is based on a makefile, you can rerun it if jobs crash or you can use "make -f $FASTHMM_DIR/lib/fbmerge.make clean" to remove the intermediate files before starting over. If you do use make to continue after jobs have crashed, you must make sure to use the same value of NPIECES or the results may not be correct.
FastHMM/FastBLAST were developed by Morgan N. Price and Y. Wayne Huang in Adam Arkin's group at Lawrence Berkeley National Lab. For more information, send e-mail to fasthmm@microbesonline.org or read the FastBLAST preprint.